|
RankVISTA
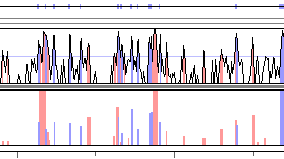 Fig. 1. A sample rankVISTA graph. |
 Fig. 2. A sample of rankVISTA regions. |
RankVISTA conservation plots depict evolutionarily conserved segments in
pairwise or multiple alignments as a bar graph, where the heights scale with
statistical significance [-log10(P-value)]. For example, a height
of 4 indicates that the probability of seeing that level of conservation by
chance in a neutrally-evolving 10-kb segment of the base sequence is less than
10-4.
RankVISTA graphs are based on the Gumby
algorithm, which estimates neutral
evolutionary rates from non-exonic regions in the multiple sequence alignment,
and then identifies local segments of any length in the alignment that evolve
more slowly than the background. The phylogenetically weighted log-odds
conservation scores of conserved segments are translated into P-values using
karlin-altschul statistics. Gumby has no window-size parameter, and no fixed
percent-identity threshold either. Since the algorithm uses a more-conserved-than
background paradigm, it can perform phylogenetic shadowing (close species) and
footprinting (distant species) with equal facility.
NOTE: Since the input alignment is its own training set, small or
grossly incomplete alignments are to be avoided:
The base sequence length should be at least 10 kb. Smaller alignments
might be tolerated, but if Gumby detects an inadequate number of aligned
positions, it will return no output.
For the p-values to be meaningful, Gumby requires a reasonably complete
alignment. Rule of thumb: the number of "N" characters and spurious gap
characters arising from missing sequence data should be less than 10% of
the total number of characters in the alignment. The relative ranking of
conserved regions by pvalue would still be meaningful if this rule were
violated. However, the p-value estimates would be systematically biased.
Gumby's sensitivity in detecting non-exonic conservation can be
increased by supplying exon annotations. The annotated regions are
masked when estimating neutral evolutionary rates, resulting in a more
accurate estimate of the background conservation level.
RankVista regions are colored according to their annotation, as seen in the figure
above. Note that RankVista coloring is based on exon annotations of all aligned
sequences, not just the one currently used as the base. Consequently, an unannotated
region in the base sequence might still be colored as an exon because of annotations
from other sequences. In another deviation from the standard scheme, RankVISTA colors
UTRs and coding exons the same, since they are treated identically by Gumby.
References:
Martin J, et al.
The sequence and analysis of duplication-rich human chromosome 16.
Nature. 2004 Dec 23;432(7020):988-94.
|